Если есть вопросы или предложения по мануалу писать в ТГ @Last_Ange1
Книга, посоветованная Александром Кликушиным. Если кому-то очень интересна тема межмолекулярных взаимодействий. В частности, глава 1.4 дает более глубокую классификацию таких взаимодействий.
Давайте эту вступительную часть разобьем на две. Сначала обсудим, что такое слабые взаимодействия, а затем, как они влияют на нашу жизнь.
Зачастую слабые взаимодействия также называют нековалентными (Noncovalent Interaction), что может вызвать путаницу, потому что в таком случае ионная связь тоже Noncovalent, хотя ее к слабым обычно не относят. Общие признаки слабых взаимодействий это отсутствие обобществления электронной пары и, удивительно, небольшая прочность связи (1—5 ккал/моль). Именно эти взаимодействия ответственны за сохранение 3D структур больших молекул (белки или нуклеиновые кислоты) и их селективное связывание. Они же отвечают за упаковку и свойства многих кристаллов и материалов. Большую роль они играют и в привычным органическом синтезе. Могут быть как меж-, так и внутримолекулярными. В конце концов, слабые взаимодействия ответственны за то что вода это жидкость, а не газ при комнатной температуре.
Все слабые взаимодействия можно поделить на 4 типа:
Электростатические (диполь — диполь):
Ионная связь (если учитываем)
Водородная связь
Галогенная связь, либо другие σ-дырочные связи, то есть нековалентное взаимодействие между ковалентно связанным атомом групп IV–VII и отрицательной частицой, например, неподеленной парой основания Льюиса или аниона. Включает область положительного электростатического потенциала, обозначенную как σ-дырка, на продолжении одной из ковалентных связей с атомом. σ-Дырка возникает из-за анизотропии распределения заряда атома
Ван-дер-Ваальсовы:
Диполь — диполь (например C=O--C=O, в ацетоне)
Диполь — наведенный диполь. Постоянный диполь поляризует неполярную молекулу, превращая ее в наведенный диполь
Наведенный диполь — наведенный диполь. Когда молекулы сближаются могут возникать временные частичные диполи, которые взаимодействуют друг с другом. Поэтому, например, гексан жидкий
π-эффекты( в общем, π-эффекты вызваны взаимодействием молекул с π-системами сопряженных молекул, таких как бензол):
π-π стэкинг
Катион — электроноизбыточная π-система или анион — электронодефицитная π-система
Диполь — π-система (более слабый вариант предыдущего)
Гидрофобный эффект. Стремление гидрофобных молекул (частей) агрегировать в водных растворах, чтобы уменьшить площадь взаимодействия с водой
Кроме того, важно помнить и об отталкивающих эффектах: В частности, стерические (отталкивание электронных оболочек) и электронные (кулоновское отталкивание).
Примеры:
Существования третичной и четвертичной структуры белка и ДНК возможно практически исключительно из-за существования слабых взаимодействий (Рисунок 1).
Рисунок 1. Строения третичной структуры и вклад слабых взаимодействий в нее
Низкие температуры кипения. Межмолекулярные взаимодействия удерживают молекулы в конденсированном состоянии. В эфире водородные связи отсутствуют, поэтому температура кипения такая низкая (Рисунок 2).
Рисунок 2. Сравнение температур кипения
Упаковка кристаллических структур. Специфические взаимодействия зачастую определяют мотив и тип упаковки в кристалле. Например, на рисунке 3, молекулы расположены "голова к голове, хвост к хвосту", то есть образуется двойной бислой: два слоя (гидрофобный и гидрофильный) из двух молекул.
Файлы расчетов (! для удобства файлы .cub переименованы, однако для работы скриптов *.vmd необходимо либо обратно назвать файлы func1.cub и func2.cub, либо в скриптах изменить имена файлов)
NCI (Noncovalent interaction) анализ состоит из двух логичных шагов. Во-первых, нам нужно определить, где в молекуле есть какие-то слабые взаимодействия (вообще неважно какого рода), а во-вторых, понять, что это за взаимодействия (насколько связывающие или разрыхляющие). Логично, не правда ли? Дальше увидим на примере как это работает, а сейчас разберемся с теорией.
На первом шаге мы воспользуемся так называемым reduced density gradient (RDG).
Рисунок 4. Вид функций Gradient norm of electron density и Reduced density gradient (RDG)
RDG выводится из плотности и ее первой производной, является фундаментальной безразмерной величиной в DFT, используемой для описания отклонения от однородного распределения электронов. В “хвостах” плотности (т.е. областях, удаленных от молекулы, в которых плотность экспоненциально уменьшается до нуля) и около ядер градиент будет иметь очень большие положительные значения. И, напротив, он будет принимать очень малые приближающиеся к нулю значения как для областей с ковалентными связями, так и с нековалентными взаимодействиями.
То же самое в виде таблички:
Рисунок 5. Таблица значений RDG и электронной плотности в разных областях молекулы. Столбцы слева направо: около ядра, около химической связи, в области слабых взаимодействий, на границе молекулы.
Ну хорошо, мы отделили ядра и края молекулы, но как разделить слабые взаимодействия и ковалентные связи. Из таблицы видно, что различия между этими областями заключаются в значениях плотности (логично: ковалентная связь прочнее, плотность больше), так что поставив нужное пороговое значение плотности можно выделить только слабые взаимодействия.
Что получилось?
Рисунок 6. Распределение градиента от электронной плотности в молекуле муравьиной кислотыРисунок 7. Распределение градиента от электронной плотности в молекуле димера муравьиной кислоты
На рисунках 6 и 7 представлены муравьиная кислота и ее димер, соответственно. Что видно на первой картинке?
область высоких значений градиента и низких значений плотности — края молекулы
область средних значений градиента и высоких значений плотности — ковалентные связи и атомы
Справа же появляются пики с малыми значениями и плотности и градиента, это и есть водородные связи в димере.
Характеристика слабых взаимодействий
Для описания слабых взаимодействий нам необходимо понять, являются ли они стабилизирующими или дестабилизирующими. Давайте посмотрим на это с точки зрения теории Бейдера (подробности обсуждается ниже). Для характеризации взаимодействия нам всего лишь нужно определить тип критической точки (КТ): (3, +1) — связывание или (3, −1) — отталкивание. Различаются эти две точки знаком второго по величине собственного значение Гессиана электронной плотности (ниже упоминается как λ2). Если λ2 > 0, то (3, +1), если наоборот, то (3, −1). Мы, конечно, не ищем КТ, но можем использовать этот подход для определения типа взаимодействия, оценивая λ2. Кроме того, сила слабого взаимодействия имеет положительную корреляцию с электронной плотностью ρ в соответствующей области. Ван-дер-Ваальсовы взаимодействия всегда имеют очень малую ρ, в отличие от областей с сильным стерическим эффектом или значительным стабилизирующим слабым взаимодействием (например, Н-связь, галогенная связь), которые всегда имеют относительно большую ρ. Таким образом мы можем определить функцию sign(λ2) ρ, а именно произведение знака λ2 и ρ. Можно использовать разные цвета для разных значений функции, что позволит легко определять эффект и силу слабого взаимодействия.
Классическая цветовая схема:
Рисунок 8. Цветовое обозначение разных значений функции sign(λ2)ρ
И давайте снова посмотрим на примеры:
Рисунок 9. Градиентные изоповерхности для (a) бицикло[2.2.2]октена, (b) разветвленного октана и димеров (c) бензола, (d) метана, (e) воды и (f) муравьиной кислоты
Особенности метода
Уровень теории, на котором были получены плотности, не важен
Отлично работает на больших молекулах (дешевый)
Определяет именно области между группами, а не отдельные пути
Результаты качественно (и почти количественно) совпадают при использование как самосогласованной плотности, так и promolecular density
Рисунок 10. ELF и радиальная плотность для атома криптона
Electron localization function (ELF) является мерой вероятности нахождения электрона в окрестности референсного электрона, расположенного в данной точке и с тем же спином. Чем сильнее локализованы электроны в области, тем более вероятно, что движение электронов ограничено внутри нее. Если электроны полностью локализованы, то их можно отличить от тех, что находятся снаружи.
Сейчас будет немного сложных фраз, впечатлительных просьба перейти к концу сегмента.
Бейдер обнаружил, что области, в которых электроны сильнее локализованы должны иметь большие величины интегрирования дырок Ферми (никакие два электрона не могут находиться в одном и том же квантовом состоянии в системе (то есть до двух электронов с разными спинами на одной орбитале)). Однако дырка Ферми является шестимерной функцией, и поэтому ее трудно изучать визуально. Беке и Эджкомб отметили, что "spherically averaged conditional same-spin pair probability density has direct correlation with the Fermi hole"... Переведем на русский.
Same-spin pair probability density (P2(r, r′)) — это плотностью вероятности одновременного нахождения двух электронов с одинаковым спином в положениях r и r′. Conditional same-spin pair probability density Pcond(r, r′) — это плотность вероятности найти электрон в некотором положении r', если референсный электрон с аналогичным спином находится с уверенностью (отсылка к принципу неопределённости Гейзенберга) в положении r.
Конец сложного сегмента.
Что мы можем вынести из этих фраз? ELF четко говорит, где локализованы электроны (не их "количество", а степень локализации), а значит мы четко можем видеть орбитали, неподеленные пары и связи.
ELF Принимает значения от нуля до единицы. Большое значение ELF означает, что электроны сильно локализованы, указывая на наличие ковалентной связи, неподеленной пары или внутренних оболочек атома. ELF = 1, соответствует полной локализации, а ELF = ½, соответствует электронному газу.
ELF часто используется для широкого спектра систем, таких как органические и неорганические малые молекулы, атомные кристаллы, координационные соединения, кластеры; а также для различных задач, таких как выявление структуры атомной оболочки, классификация химических связей, проверка Charge-shift bonds (что это, можно почитать тут), изучение ароматичности.
Рисунок 11. DORI для (a) димера воды, (b) димера аммиака, (c) димера метана, (d) димера муравьиной кислоты, (e) π-stacked димера бензола, (f) Т-образного димера бензола, (g) π-stacked аденин–тимина, (h) аденин–тимина, связанного водородной связью. Цвет изоповерхности соответствует sign(λ2)ρ(r) в диапазоне от -0,02 (красный) до 0,02 (синий)
Мы уже посмотрели на ряд функций, описывающих структуру молекулы тем или иным способ. Однако что будет если их объединить? Мы не первые, кто задается таким вопросом. В частности, можно объединить ELF и RDG для исследования ковалентных и слабых взаимодействий (сам по себе RDG тоже можно для этого использовать, но оптимальные значения изоповерхностей для разных взаимодействий разнятся, поэтому такой подход очень сложен и не используется ). Объединяющей их функцией стал DORI. При правильных значениях изоповерхности (isovalue) оба типа взаимодействий можно изобразить на изоповерхности DORI, ровно как и sign(λ2) ρ, который облегчает анализ характера взаимодействий. Индекс принимает значения от 0 до 1
В настоящий момент, DORI не рекомендуют к использованию из-за двух причин: (1) Получение DORI сложно, и для его вычисления требуется производная второго порядка электронной плотности, что сложнее реализовать и посчитать, чем RDG. (2) Графический вид изоповерхности DORI часто неудовлетворителен, а в некоторых регионах DORI получается численно шумным. Вместо него предлагают использовать Interaction Region Indicator (IRI), созданный автором Multiwfn. Его мы и обсудим следующим.
Interaction Region Indicator это немного измененный RDG. Он делает то же, что и DORi, но считается проще и выглядит красивее. По существу, IRI это градиентная норма электронной плотности (Рисунок 4) , взвешенная (поделенная) по масштабированной (то есть в некоторой степени) электронной плотности. Значение а было найдено экспериментально при анализе большого числа систем, при а=1 функция отличается от RDG только коэффициентом. При правильном выборе значений изоповерхности индикатор позволяет изображать регионы с разными по силе взаимодействиями на одной картинке. К IRI можно точно также добавить sign(λ2) ρ для отображения природы показных взаимодействий. Цветовая схема, физический и химический смысл изображен на Рисунке 8.
Не буду переводить здесь перевод всей статьи, а лишь выделю ключевые моменты
При сравнение с RDG хорошо видна возможность аккуратного изучения взаимодействий всех типов на одной картинке с одним значением изоповерхности
Нет настолько большой чувствительности к выбору isovalue, как у DORI, IRI просто красивее (меньше шумов, фрагментации), лучше разделение между регионами ковалентных связей
IRI в некоторых случаях позволяет определить взаимодействия невидимые даже для QTAIM
IRI удобно использовать для изучения химической реакции, потому что можно визуализировать плавный переход от нековалентного взаимодействия к химической связи
Рисунок 13. 1. Карта изоповерхностей димера фенола RDG = 0.5, RDG = 0.25 и IRI = 1.1. 2. Карта изоповерхностей двух молекул диамантана IRI = 1.0, DORI = 0.96. 3. Карты Ni(NH3)2(OH)2: (а) График IRI vs sign(λ2)ρ(r), (b) Карта изоповерхности IRI = 1.0, (c) Карта градиентных линий с контурными линиями ρ на молекулярной плоскости. Синие точки соответствуют BCPs, коричневые линии представляют bond paths. 4. Изменения в карте изоповерхности IRI = 0.95 при реакции Дильса-Альдера между цис-1,3-бутадиеном и этиленом.
Рекомендую прочитать оригинальную статью, а также использовать IRI в своих исследованиях. Пример использования, как обычно, ниже.
Теория AIM, предложенная Бейдером описывает систему через атомы и связи, положения, которых можно найти из электронной плотности. Представляются атомы и связи как критические точки (точки экстремумов электронной плотности). Из вышесказанного можно сделать вывод, что система определяется числом и типом критических точек. а также силовыми линиями, которые начинаются и кончаются в этих точках.
Есть 4 типа критических точки. Они определяются двумя значениями (w, s). w — размерность пространства в котором построена электронная плотность (как правило 3). s — сумма знаков собственных значений Гессиана электронной плотности. Знак собственного значения указывает, уменьшается (−) или увеличивается (+) электронная плотность при смещении по данному координатному направлению. Сейчас рассмотрим на примерах.
Если точка является максимумом по какому-то направлению, то при смещении по данному координатному направлению ЭП будет уменьшаться и знак точки по этому направлению будет "-". И наоборот.
Типы критических точек
(3, −3) — ядро; все три собственных значения Гессиана отрицательны, то есть это локальный максимум, во всех трех направлениях электронная плотность уменьшается При анализе электронной плотности и для тяжелых атомов положение (3, −3) почти идентично положению ядра, следовательно, (3, −3) также называется ядерной критической точкой (NCP). Обычно их количество равно числу атомов (есть случаи, где это не так, но мы это не разбираем).
(3, −1) — связь; два собственных значения Гессиана отрицательны, и это седловая точка второго порядка. В двух направлениях ЭП уменьшается, в одном растет. При анализе электронной плотности критическая точка (3, −1) обычно появляется между связывающими атомными парами, потому обычно называется критической точкой связи (BCP). Важность значений функций реального пространства (real space function) в BCP сложно переоценить. Именно они (обсуждается чуть дальше) определяют тип и прочность связи. "Функциями реального пространства" в Multiwfn называются функции, переменными которых являются координаты трехмерного пространства данной системы.
(3, +1) — кольцо; только одно собственное значение Гессиана отрицательно, и это седловая точка первого порядка (как переходное состояние на поверхности потенциальной энергии). ЭП уменьшается в одном направление, а в двух растет. При анализе электронной плотности точка (3, +1) обычно появляется в центре кольца и отражает стерический фактор, поэтому (3, +1) часто называют критической точкой кольца (RCP).
(3, +3) — cage (полость); ни одно из собственных значений Гессиана не является отрицательным, это локальный минимум. При анализе электронной плотности (3, +3) обычно появляется в центре системы клеток (например, пирамидальная молекула P4), поэтому ее часто называют критической точкой клетки (CCP).
Для системы с конечным числом атомов должен выполняться Принцип Пуанкаре — Хопфа n(3, −3) − n(3, −1) + n(3, +1) − n(3, +3) = 1, где n(3, x) — число КТ указанного в скобках типа.
Рисунок 14. Молекулярный граф
Параметры критических точек
ρ – величина электронной плотности
∇2𝜌- величина Лапласиана электронной плотности
Рисунок 15. Лапласиан электронной плотности
ε – эллиптичность электронной плотности
V(r) – плотность потенциальной энергии
Величина электронной плотности и Лапласиана электронной плотности
Напрямую связаны с энергией химической связи. Это справедливо не только для ковалентных связей, но и для нековалентных, например, водородных связей. Величина Лапласиана электронной плотности коррелирует с энергией связи, а знак Лапласиана электронной плотности указывает на тип химической связи.
∇2𝜌 < 0: наблюдается концентрация электронной плотности в пространстве между атомами — ковалентная связь (доминирует орбитальная составляющая связи).
∇2𝜌 > 0 – наблюдается разрежение электронной плотности в пространстве между атомами — ионная связь (доминирует электростатическая составляющая связи).
Эллиптичность электронной плотности
Является количественной характеристикой несферичности распределения электронной плотности на химических связях.
ε = 0 для σ-связей (например С−С в этане)
ε > 0 для двойных связей, содержащих π-компоненту (например С=С в этилене)
ε = 0 для тройных связей, содержащих π-компоненту (например С−С в ацетилене)
Величина эллиптичности коррелирует с энергией пи-связывания.
Плотность потенциальной энергии
Связана с энергией межмолекулярных взаимодействий.
Bond paths
Путь c максимальным градиентом, соединяющий BCP и связанные с ним два локальных максимума плотности, называется “Bond path" ("путь связи"). Он показывает путь взаимодействия атомов для всех видов связи. Совокупность путей связи известна как молекулярный граф, который обеспечивает однозначное определение молекулярной структуры. Траектория соединения может быть прямой или кривой.
Multiwfn
Файлы расчетов (! для удобство файлы .cub переименованы, однако для работы скриптов *.vmd необходимо либо обратно назвать файлы func1.cub и func2.cub, либо в скриптах изменить имена файлов)
"Multiwfn is an extremely powerful program for realizing electronic wavefunction analysis, which is a key ingredient of quantum chemistry. Multiwfn is free, open-source, high-efficient, very user-friendly and flexible, it supports almost all of the most important wavefunction analysis methods."
Если кратко, Multiwfn это очень полезная программа для анализа волновой функции. Давайте учиться ей пользоваться.
3. Все! Теперь настроем использование памяти и переменные (как сделать, чтобы Multiwfn можно было вызвать откуда угодно по его названию). Для этого нам нужно открыть файл ~/.bashrc
nano ~/.bashrc
// если не работает nano, давайте поставим ее //
conda install -c conda-forge nano
4. Пишем в него
export KMP_STACKSIZE=200M
ulimit -s unlimited
export Multiwfnpath=/путь к Multiwfn/ (например /home/chaliv/bin/Multiwfn/Multiwfn_3.8_dev_bin_Linux)
export PATH=$PATH:/путь к Multiwfn/ (например /home/chaliv/bin/Multiwfn/Multiwfn_3.8_dev_bin_Linux)
5. Чтобы изменения в ~/.bashrc вступили в силу, используем source ~/.bashrc. Затем делаем файл Multiwfn исполняемым
chmod +x /относительный или абсолютный путь к Multiwfn/Multiwfn(например /bin/Multiwfn/Multiwfn_3.8_dev_bin_Linux/Multiwfn)
6. Наконец изменим настройки в setting.ini, который находится там же, где и исполняемый файл
nthreads = число, на сколько потоков вы хотите параллелить расчеты.
Число зависит от того, где вы считаете. Если на своей машине или нашем сервере, где сейчас ничего стоит, можно ставить, сколько угодно. Если вы считаете на курчатнике, то скорее всего, вы не ставите расчет в multiwfn на какую расчетную ноду, а считаете на головной. В таком случае лучше не занимать всю ноду и можно поставить, например, 4.
Windows
1. Скачать отсюда архив "Multiwfn_3.8_dev_bin_Win64.rar" распаковать и все. Буквально. Иногда необходимо сделать файл Multiwfn исполняемым (Свойства — Безопасность — Изменить). Готово! Тут даже есть графический интерфейс.
Практическая часть
Как получить плотность
Для всех методов нужна электронная плотность, разберёмся как ее получить из Gaussian и Orca.
Gaussian
Плотность получаем из .fchk или .wfn файла. Как их получить?
Давайте разберем два варианта. Для первого вам просто нужно превратить .chk в .fchk утилитой Gaussian formchk.
Второй вариант сложнее и обычно бесполезнее, так как wfn несет меньше информации, чем .fchk, но давайте рассмотрим его тоже
Для получения wfn нам нужно специально попросить это в Инпут-файле. Из необычного только output=wfn, просим gaussian вывести волновую функцию в отдельный файл.
Готово! Мы получили .wfn файл.
Orca
Орка создает много разных файлов и часть их них может быть использована для работы с Multiwfn, но не все типы файлов созданы равными. Лучше всего использовать .gbw, однако сам по себе Multiwfn принять его не может, его необходимо преобразовать в .molden файл, с помощью утилиты орки, orca_2mkl (путь на hpc4 /s/ls4/groups/g0130/lsvvt/motorca504/orca/orca_2mkl)
orca_2mkl file -molden
После этой процедуры мы получим .mkl файл, это на самом деле и есть .molden и уже его можно давать Multiwfn
Для некоторых функций неописанных в этом мануале могут понадобиться другие типы файлов или не каждый из описанный подойдет. Для проверки стоит обратиться к параграфу 2.5 мануала по MultiWFN (Input files and wavefunction types)
Запускаем Multiwfn (из любого места, если он прописан в PATH).
Multiwfn ./manual_weak.wfn или Multiwfn ./manual_weak.fchk
Теперь надо пройти по меню Multifwn нажимая на цифры:
20 // Visual study of weak interaction (логично, читай название мануала)
1 // NCI analysis (еще более логично. Там есть пункт 2 — NCI based on promolecular theory. Это нужно для больших систем, где мы считаем электронную плотность, как суперпозицию электронных плотностей молекул. Используйте этот вариант, если, например, считаете кусок ДНК)
2 // Medium quality grid (выбирайте грид в зависимости от размера системы)
3 // Output cube files to func1.cub and func2.cub in current folder (сохранить cub файлы для визуализации. В них лежит посчитанная нами grid data для функций sign(λ2) ρ и RDG, то есть форма и цвет поверхностей)
VMD
Теперь самый важный этап, будем рисовать картинки. Для этого нам нужен VMD.
Скачиваем отсюда и устанавливаем. Все работает, но VMD просит класть файлы визуализации в одну с ним папку, а, как справедливо заметили мои коллеги, класть файлы в системную библиотеку неправильно (а может быть и невозможно, если вы не админ).
Чтобы исправить это недоразумение, мы можем поступить тремя варинатами. Мы обсудим все. Делайте как вам больше нравится.
1) Надо добавить VMD в PATH (переменная окружения, в которой лежит пути к разным папкам, которые мы можем запускать откуда угодно). В Linux это делается через редактирование файла ~/.bashrc (как это сделано выше для Multiwfn), в Windows можно воспользоваться графическим интерфейсом.
Открываем панель управления (Control panel)
В поиске (справа сверху) пишем "Изменение системных переменных среды"
Жмем на "Изменение системных переменных среды"
Вкладка Дополнительно — Переменные среды...
В системных переменных ищем PATH
Изменить
В новом окне — Обзор
Выбираем папку с VMD и жмем ОК
ОК в окне с PATH
OK в окне переменные среды
ОК в окне свойства системы
Теперь можно вызывать vmd откуда угодно (через консоль), а также можно давать ему файлы, лежащие в любом месте (для этого нужно в консоли зайти в папку с файлами и открыть VMD, вызвав vmd.exe). Просто кладем скрипт для визуализации в любую папку, к нему же кладем файлы для визуализации (.cub .pdb и т.д.)
1.1) Есть вариант добавления VMD в path для конкретной сессии (отдельного окна командной строки). Для этого в командной сроке надо прописать следующую команду:
path "путь\до\VMD"
Она добавит VMD в локальный path для этой конкретной сессии командной строки. То есть это нужно делать каждый раз при запуске новой cmd
2) Альтернативный вариант — в файлах .vmd с командами для визуализации (RDGfill.vmd, AIM.vmd ...) добавить полный путь к загружаемый молекулам. Например:
mol new "C:/notcyrylic/multiwfn_aimall/manual/func1.cub"
Важные моменты:
В пути не должно быть русских букв
Путь должен быть в кавычках и с прямым слэшами (/), даже если работа ведётся в WIndows
3) Самый простой способ, писать в командой строке "start VMD", программа откроется и вы сможете пользоваться, ей как в первом варианте
Какой путь вы бы ни выбрали, все готово и теперь:
Копируем файлы (func1.cub и func2.cub) и файл RDGfill.vmd из папки examples установочной директории Multiwfn в любую папку
Запускаем VMD (через консоль и из этой папки)
В графическом интерфейсе жмем "File" — "Load Visualization State…", выбираем RDGfill.vmd (либо прописываем в консоли этот файл)
Еще можно передать файл .vmd через аргумент -е при запуске VMD, например
VMD -e ./RDGfill.vmd
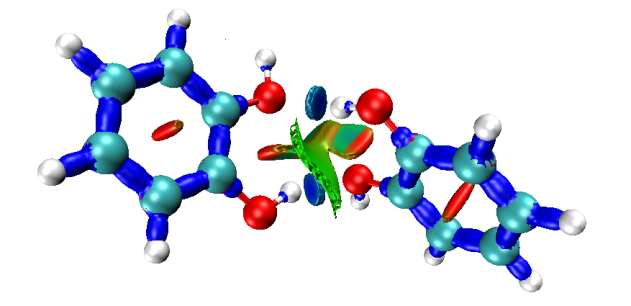
Давайте посмотрим внимательно на получившуюся картинку:
Рисунок 16. NCI анализ молекулы димера пирокатехина
Сразу бросается в глаза два синих диска, отвечающих за связи O···OH связь, как мы все знаем — это "сильные" связывающие взаимодействия. Похожее взаимодействие существует и внутри одной молекулы, однако оно слабее и на рисунке отражено голубым. На концах большой изоповерхности присутствуют оранжевые участки, символизирующие отталкивание двух кислородов, а центральная зеленая часть ответственна за ван-дер-ваальсовы взаимодействия. В центре обоих бензолов есть изоповерхность красного цвета, что вызвано стерическим отталкиванием.
NCI + ELF
5 часть видео из шапки NCI
В VMD Рисуем NCI как показано раньше.
Считаем ELF
5 // Output and plot specific property within a spatial region (calc. grid data) (Нам оно и нужно)
9 // Electron Localization Function (ELF) (комментарии излишни)
2 // Medium quality grid, covering whole system, about 512000 points in total (возьмем среднее количество точек)
2 // Export data to a Gaussian-type cube file in current folder (экспорт куб-файла c точками, которые нужно отобразить)
Переносим ELF.cub в VMD. Можно просто перетянуть файлик в VMD main (окно с панелью команд). Если не получается перетянуть, то открываем файл через "File" — "New Molecule…"
Нажимаем на Display и ставим галочку в поле Light 3
Настраиваем оторажение
Заходим в Graphics — representation.
Выбираем файл ELF
Drawing method — isosurface
Настройте isovalue (около 0.8 будет нормально)
Material GlassBubble
Coloring method — ColorID — 3
Можно немного увеличить значение isovalue для NCI, чтобы она не терялась на фоне ELF
Готово!
Рисунок 17. NCI + ELF
Что можно увидеть тут? Например, что пара кислорода в водородной связи ориентирована на водород.
ELF напрямую не настолько полезен для изучения слабых взаимодействий, поэтому обсуждения других вариантов его представления мы отложим.
NCI + AIM information
в 4 части видео из шапки есть алгоритм с использование скриптов из папки examples/scripts. Тут я покажу ручной вариант, чтобы вы понимали, как скрипты работают.
То что мы получили уже позволяет многое сказать об изучаемой молекуле, но давайте пойдем немного дальше и посмотрим одновременно на NCI и критические точки из теории Бейдера (про это я тоже напишу, но позже).
Как нам это сделать? Начнем с части в Multiwfn:
Возвращаемся обратно в меню
2 // Topology analysis (логично, теория Бейдера это как раз он)
Ищем критические точки
2 // Search CPs from nuclear positions
3 // Search CPs from midpoint of atomic pairs
4 // Search CPs from triangle center of three atoms (не факт, что это вам надо)
5 // Search CPs from pyramid center of four atoms (не факт, что это вам надо)
8 // Generate bond path (соединяем CP ядер и связей)
Экспорт CP
-4 // Modify or export CPs
6 // Export CPs as CPs.pdb in current folder (файл со списком критических точек, их параметрами и координатами)
0 // Return
Экспорт путей
-5 // Modify or print detail or export paths
6 // Export paths as paths.pdb in current folder (файл, состоящий из блоков, каждый блок описывает один путь)
Закрываем Multiwfn
Переходим в VMD.
Копируем оба созданных .pdb и куб файлы NCI в нужную вам папку
Из папки Multiwfn examples/scripts копируем скрипт AIM.vmd
Загружаем AIM.vmd и RDGfill.vmd по очереди, как в прошлый раз
Все получилось. Если получились как на рисунке 18, поставьте значение isovalue (смотри часть про NCI+ELF) на 0.5 (или поварьируйте)
Рисунок 18. NCI с AIM при неправильном isovalueРисунок 19. NCI с путями и критическими точками из теории Бейдера
Получившиеся критические точки вместе с изоповерхностью позволяют нам более полно описать слабые взаимодействия в системе. Например, теперь мы однозначно видим взаимодействие кислород — кислород, а по изоповерхности определяем что оно относится к ван-дер-ваальсовым.
По итогу хочется сказать, что NCI достаточной простой, но в то же время очень полезный метод.
DORI
Параграф 4.20.5 в мануале
Как было сказано выше, IRI делает все то же самое, но лучше. Однако давайте сами в этом убедимся, да и уметь делать DORI лишним не будет. Ход работы, такой же как раньше. Нажимаем на цифры в Multiwfn, скриптом делаем картинку в VMD.
Multiwfn:
Запускаем "Multiwfn manual_weak.wfn ", оказываемся в меню
Делаем DORI
20 // Visual study of weak interaction
5 // DORI analysis
3 // High quality grid (опять-таки зависит от размеры вашей молекулы, у нас маленькая система, так что grid большой можно)
3 // Export cube file (получили cub)
VMD:
Запускаем VMD
Кладем в папку оба .cube файл и DORIfil.vmd (лежит в Multiwfn_3.8_dev_bin_Linux/examples/ DORIfil.vmd)
В графическом интерфейсе жмем "File" — "Load Visualization State…", выбираем Dorifill.vmd (либо прописываем в консоли этот файл)
Готово Рисунок 20. DORI молекулы димера пирокатехина Результат очень похож на NCI, однако как и обещалось, видны как слабые взаимодействия, так и ковалентные связи. Слабые взаимодействия были разобраны выше, не будем повторяться. Картинка выглядит неплохо, но давайте сначала посмотрим на IRI и потом сравним
IRI
Параграф 4.20.4 в мануале
Изоповерхность
Multiwfn:
Запускаем "Multiwfn manual_weak.wfn ", оказываемся в меню
Делаем IRI
20 // Visual study of weak interaction
4 // IRI analysis
3 // High quality grid
3 // Export cube file
VMD:
Запускаем VMD
Кладем в папку оба .cube файл и IRIfil.vmd (лежит в Multiwfn_3.8_dev_bin_Linux/examples/ IRIfil.vmd)
В графическом интерфейсе жмем "File" — "Load Visualization State…", выбираем IRIfill.vmd (либо прописываем в консоли этот файл)
Готово
Рисунок 21. IRI молекулы димера пирокатехина
Как можно видеть репрезентация слабых взаимодействии практически не отличается от таковой в DORI, однако ковалентные связи отдалены одна от другой и изображены намного более аккуратно. Считается IRI тоже быстрее (разница примерно в 2 раза, на больших молекулах будет заметно) Из написанного в разделе с теорией и приведенных сейчас картинок можно смело говорить, что IRI является более оптимальным инструментом для оценки и визуализации слабых взаимодействий.
Плоскость
Иногда может быть полезно построить карту IRI на плоскости, чтобы выявить области взаимодействия в определенной плоскости. Вам наверное уже надоел пирокатехина. поэтому давайте возьмем молекулу из примера в мануале (examples\GC.wfn есть у меня в архиве) (так как мы рисуем в плоскости, нужно будет осознавать какие атомы лежат в этой плоскости, если у вас очень не плоская молекула, то ничего разумного вы, скорее всего, не получите (я пробовал)). Со всем приведенным ниже можно и иногда нужно экспериментировать. Менять значения, цветовые палитры и так далее.
Multiwfn:
В файле settings.ini поставить iuserfunc = 99, чтобы user function была IRI
Запускаем Multiwfn GC.wfn
Рисуем
4 // Output and plot specific property in a plane (мы и хотим в плоскости)
100 //User-defined function (iuserfunc= 99) (то что мы ставили в settings.ini)
1 // Color-filled map (означает тип карты, в нашем случае цветная)
[Press ENTER button to use default number of grids] (применяем дефолтное количество гридов, не понравится можно поменять)
0 // Set extension distance for plane type 1~5 (как я понял, это то насколько широкая у нас плоскость)
1 // 1 Bohr (ставим ширину)
1 // XY plane (в какой плоскости будет наша плоскость)
0 // Z=0 (какое Z)
Close the graph and then input (ждем пока отрисуется график и потом его закрываем)
19 // Set color transition (это по идее цветовая палитра)
2 // Reversed rainbow (можно экспрементировать)
1 // Set lower&upper limit of color scale (по факту, это какие краски мы включаем в картинку из радуги)
0,2
4 // Enable showing atom labels and reference point
1 // Red (каким цветом красим атомы)
8 // Enable showing bonds
14 // Brown (каким цветом красим связи)
-1 // Plot again (рисуем и ждем)
0 // Save the graph to a graphical file in current folder (сохраняем в папку)
Красота!
Рисунок 22. IRI в плоскости
Оранжевая и зеленая области (IRI < 1,0) на этой карте четко показывают области, где происходит заметное взаимодействие химических связей и слабое взаимодействие. Области с IRI >1,0 имеют либо большой градиент электронной плотности, либо незначительную электронную плотность, они не представляют химического интереса.
ESP
Multiwfn:
Запускаем "Multiwfn .fchk ", оказываемся в меню
Делаем ESP
Сначала получаем электронную плотность в cub файла
5 (выдать данные в области)
1 (конкретно электронная плотность)
3 (количество точек электронной плотности/ разрешение/ grid, я взял “High quality”)
2 (выдать ЭП в виде .cub файла – файлы данного формата считаются классическим представлением grid)
0 (возвращаемся в главное меню)
Теперь надо получит esp нарисованное по grid из электронной плотности (считает ESP на поверхности электронной плотности)
5 (выдать данные в области)
12 (esp)
8 (grid из внешнего файла, в нашем случае .cub плотности)
./density.cub (прописать путь к .cub плотности из пункта 2)
2 (выдать esp в виде .cub файла)
Paraview:
Загрузить и отрисовать в Paraview, воспользовавшись мануалом Тимофея
Пример ESP ниже
Создатели
Чалый Василий. СоздательАлександр Кликушин. Самый заинтересованный редактор. Внес неоценимый вклад в описание самих по себе слабых взаимодействий, а его советы не только помогли мне сделать этот мануал лучше, но и подтолкнули к дальнейшему его развитию.Игорь Мезенцев. Еще один человек, напомнивший мне о важности этого мануала. Редактор, который не просто оставил несколько вопросов, но и помог на них ответить, там где моей компетенции не всегда хватало.
Тимофей Лосев. Самый дотошный редактор. Настолько выверенный и аккуратный мануал стал возможен во многом благодаря нему. Мной были переписаны и добавлены целые куски текста по его советам. Отсутствие орфографических и пунктуационных ошибок также в большой степени является его заслугой.Ксения Цыкина. Редактор, который на момент публикации этой картинки еще сохранял человеческие черты (которые я и другие редакторы уже потеряли) и мог посмотреть на мануал взглядом человека, для которого теоретическая химия была еще совсем новой областью. Рецензия Ксения была очень полезна, я смог понять, каким местах нужно уделить дополнительное внимание и расширить (добавить) объяснение.



.png)




![Градиентные изоповерхности для (a) бицикло[2.2.2]октена, (b) разветвленного октана и димеров (c) бензола, (d) метана, (e) воды и (f) муравьиной кислоты](/wiki/File:Rdg_examples.png)